






Dr. Felix Stelter
PD Dr. med. habil. Felix Stelter ist Ärztlicher Leiter der Labor Augsburg MVZ GmbH und Sprecher der AG QM des ALM e.V.



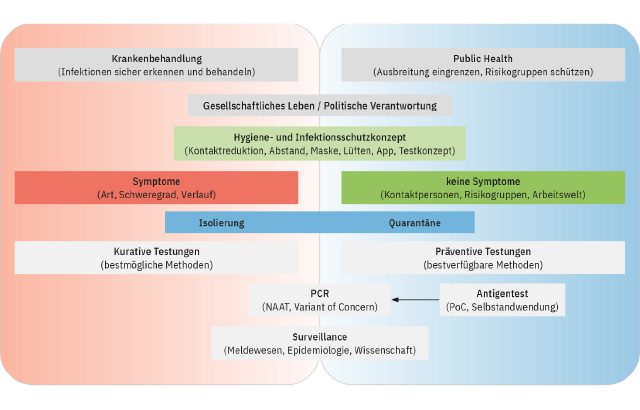
COVID–19 ist eine globale und bedrohliche Infektionserkrankung — neben vielen anderen
COVID-19 beherrscht seit zwei Jahren weltweit das gesellschaftliche Miteinander. Doch was kommt danach?
Die Datenerhebung des ALM e.V. zur SARS-CoV-2-PCR-Testung: „Woche für Woche — ein Kraftakt für alle“
Ein Interview mit Uli Früh, der seit Beginn der Pandemie die Abfragen aus über 180 Laboren koordiniert und auswertet
Intensivierung der praktischen Ausbildung in den Laboren bei ungeklärter Finanzierung
Die gesetzlichen Grundlagen der MTLA-Ausbildung ab 2023 leiden an einer massiven Finanzierungslücke
FBREK: Gefährdung der fachärztlichen humangenetischen Versorgung
Die Zertifizierung von FBREK bedeutet keine Verbesserung der Versorgung betroffener Familien – im Gegenteil.
Labore: Plötzlich im Fokus der Öffentlichkeit
Erfahrungen aus zwei Jahren PR-Arbeit während der Pandemie
10.000 Follower und 300 Tweets später — ein Resümee
Anfang 2020 folgten dem ALM e. V. auf Twitter 35 Personen, zwei Jahre später sind es 10.870